Genomics of microbial assemblage in Western Pacific Ocean
 The microbial community in the interior of oceans are still mostly unexplored. In this project, we selected South China Sea as a studying target in the survey of microbial community in the interior of ocean.
The microbial community in the interior of oceans are still mostly unexplored. In this project, we selected South China Sea as a studying target in the survey of microbial community in the interior of ocean.  We adopt the next generation sequencing technique to look at potential metabolic profiles at varying depths. My colleagues Dr. Shiah from Research Center for Environmental Change collected samples from four depths for us.
We adopt the next generation sequencing technique to look at potential metabolic profiles at varying depths. My colleagues Dr. Shiah from Research Center for Environmental Change collected samples from four depths for us.  We have completed genomic sequencing experiments and is now working on computational analyses. Chiang et al., Diversity and composition of bacteria in the interior of the South China Sea, a list of the taxonomic affiliation of each selected band sequence from the DGGE gel. LINK International Jurnal of Molecular Sciences (2014) The review paper: Marine Microbial Metagenomics: from individual to the environments. Microbes are the most abundant biological entities on earth, therefore, studying them is important for understanding their roles in global ecology. The science of metagenomics is a relatively young field of research that has enjoyed significant effort since its inception in 1998. Studies using next-generation sequencing techniques on single genomes and collections of genomes have not only led to novel insights into microbial genomics, but also revealed a close association between environmental niches and genome evolution. Herein, we review studies investigating microbial genomics (largely in the marine ecosystem) at the individual and community levels to summarize our current understanding of microbial ecology in the environment.
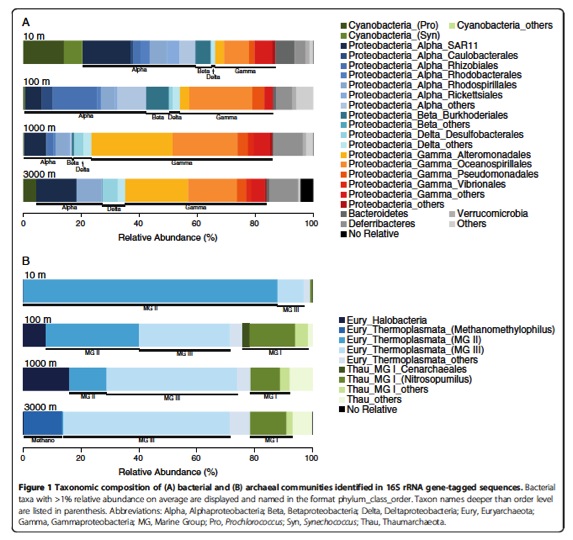
We have completed genomic sequencing experiments and is now working on computational analyses. Chiang et al., Diversity and composition of bacteria in the interior of the South China Sea, a list of the taxonomic affiliation of each selected band sequence from the DGGE gel. LINK International Jurnal of Molecular Sciences (2014) The review paper: Marine Microbial Metagenomics: from individual to the environments. Microbes are the most abundant biological entities on earth, therefore, studying them is important for understanding their roles in global ecology. The science of metagenomics is a relatively young field of research that has enjoyed significant effort since its inception in 1998. Studies using next-generation sequencing techniques on single genomes and collections of genomes have not only led to novel insights into microbial genomics, but also revealed a close association between environmental niches and genome evolution. Herein, we review studies investigating microbial genomics (largely in the marine ecosystem) at the individual and community levels to summarize our current understanding of microbial ecology in the environment.  BMC Bioinformatics Ching-Hung Tseng, Pei-Wen Chiang, Hung-Chun Lai, Fuh-Kwo Shiah, Ting-Chang Hsu, Yi-Lung Chen, Liang-Saw Wen, Chun-Mao Tseng, Wung-Yang Shieh, Isaam Saeed, Saman Halgamuge, Sen-Lin Tang, 2015, Prokaryotic assemblages and metagenomes in pelagic zones of the South China Sea, BMC Genomics, 16:219 doi:10.1186/s12864-015-1434-3 (With a Flag "Highly accessed") Abstract Background: Prokaryotic microbes, the most abundant organisms in the ocean, are remarkably diverse. Despite numerous studies of marine prokaryotes, the zonation of their communities in pelagic zones has been poorly delineated. By exploiting the persistent stratification of the South China Sea (SCS), we performed a 2-year, large spatial scale (10, 100, 1000, and 3000 m) survey, which included a pilot study in 2006 and comprehensive sampling in 2007, to investigate the biological zonation of bacteria and archaea using 16S rRNA tag and shotgun metagenome sequencing. Results: Alphaproteobacteria dominated the bacterial community in the surface SCS, where the abundance of Betaproteobacteria was seemingly associated with climatic activity. Gammaproteobacteria thrived in the deep SCS, where a noticeable amount of Cyanobacteria were also detected. Marine Groups II and III Euryarchaeota were predominant in the archaeal communities in the surface and deep SCS, respectively. Bacterial diversity was higher than archaeal diversity at all sampling depths in the SCS, and peaked at mid-depths, agreeing with the diversity pattern found in global water columns. Metagenomic analysis not only showed differential %GC values and genome sizes between the surface and deep SCS, but also demonstrated depth-dependent metabolic potentials, such as cobalamin biosynthesis at 10 m, osmoregulation at 100 m, signal transduction at 1000 m, and plasmid and phage replication at 3000 m. When compared with other oceans, urease at 10 m and both exonuclease and permease at 3000 m were more abundant in the SCS. Finally, enriched genes associated with nutrient assimilation in the sea surface and transposase in the deep-sea metagenomes exemplified the functional zonation in global oceans. Conclusions: Prokaryotic communities in the SCS stratified with depth, with maximal bacterial diversity at mid-depth, in accordance with global water columns. The SCS had functional zonation among depths and endemically enriched metabolic potentials at the study site, in contrast to other oceans.
BMC Bioinformatics Ching-Hung Tseng, Pei-Wen Chiang, Hung-Chun Lai, Fuh-Kwo Shiah, Ting-Chang Hsu, Yi-Lung Chen, Liang-Saw Wen, Chun-Mao Tseng, Wung-Yang Shieh, Isaam Saeed, Saman Halgamuge, Sen-Lin Tang, 2015, Prokaryotic assemblages and metagenomes in pelagic zones of the South China Sea, BMC Genomics, 16:219 doi:10.1186/s12864-015-1434-3 (With a Flag "Highly accessed") Abstract Background: Prokaryotic microbes, the most abundant organisms in the ocean, are remarkably diverse. Despite numerous studies of marine prokaryotes, the zonation of their communities in pelagic zones has been poorly delineated. By exploiting the persistent stratification of the South China Sea (SCS), we performed a 2-year, large spatial scale (10, 100, 1000, and 3000 m) survey, which included a pilot study in 2006 and comprehensive sampling in 2007, to investigate the biological zonation of bacteria and archaea using 16S rRNA tag and shotgun metagenome sequencing. Results: Alphaproteobacteria dominated the bacterial community in the surface SCS, where the abundance of Betaproteobacteria was seemingly associated with climatic activity. Gammaproteobacteria thrived in the deep SCS, where a noticeable amount of Cyanobacteria were also detected. Marine Groups II and III Euryarchaeota were predominant in the archaeal communities in the surface and deep SCS, respectively. Bacterial diversity was higher than archaeal diversity at all sampling depths in the SCS, and peaked at mid-depths, agreeing with the diversity pattern found in global water columns. Metagenomic analysis not only showed differential %GC values and genome sizes between the surface and deep SCS, but also demonstrated depth-dependent metabolic potentials, such as cobalamin biosynthesis at 10 m, osmoregulation at 100 m, signal transduction at 1000 m, and plasmid and phage replication at 3000 m. When compared with other oceans, urease at 10 m and both exonuclease and permease at 3000 m were more abundant in the SCS. Finally, enriched genes associated with nutrient assimilation in the sea surface and transposase in the deep-sea metagenomes exemplified the functional zonation in global oceans. Conclusions: Prokaryotic communities in the SCS stratified with depth, with maximal bacterial diversity at mid-depth, in accordance with global water columns. The SCS had functional zonation among depths and endemically enriched metabolic potentials at the study site, in contrast to other oceans.